4. Einschätzung verschiedener Anwendungsfälle
Der Einsatz von KI in der medizinischen Dokumentation wirft neue regulatorische Fragen auf: Ab wann gilt eine Software als Medizinprodukt – insbesondere bei grossen Sprachmodellen? Das Kapitel beleuchtet Abgrenzungskriterien, rechtliche Unsicherheiten und zentrale Anwendungsfälle.

Einleitung
Bei der Qualifikation von KI-basierter Software zur Erstellung von Medizinberichten als Medizinprodukte steht die Frage im Vordergrund, ob die Software die Diagnose oder die Wahrnehmung der Ärztinnen und Ärzte beeinflussen kann. Wichtig ist hier der Verwendungszweck. Wenn die Software beispielsweise medizinische Daten für eine Krankenhausstatistik auswertet oder für die Vergütung von Krankenkassen umformatiert, dann erfüllt sie keine medizinischen Funktionen.
Auch wenn eine Software grundsätzlich unter die Definition eines Medizinprodukts fällt, gilt sie nicht als solches, wenn sie ausschliesslich Funktionen wie Speicherung, Archivierung, verlustfreie Kompression, einfache Suche oder Kommunikation von Daten übernimmt. Diese funktionalen Ausnahmen sind im geltenden Recht definiert. Sie lassen sich jedoch nicht eindeutig auf KI-Systeme wie LLMs übertragen – insbesondere im Hinblick auf deren Kommunikationsfunktionen oder semantische Suchfähigkeiten. Hier besteht derzeit keine einheitliche Auslegung durch Zulassungsstellen und Aufsichtsbehörden.
LLMs bringen ganz neue Risiken mit sich, und gängige regulatorische Konzepte wie Reproduzierbarkeit, algorithmische Transparenz, generell akzeptierte Metriken für Genauigkeit und Robustheit sind nur beschränkt anwendbar. Regulierungsbehörden in verschiedenen Ländern sind momentan dabei, Konzepte für die Klassifizierung und Dokumentation von solchen Systemen zu erarbeiten. Es wird aber vermutlich noch einige Jahre dauern, bis sich ein allgemeiner Konsens zu diesen Themen herausschält und erste Leitlinien und Leiturteile von Gerichten vorliegen.
Verschiedene KI-Anwendungsfälle in der medizinischen Dokumentation
Die nachfolgenden Szenarien unterscheiden sich in ihrer technischen Umsetzung und regulatorischen Einordnung. KI-gestützte Lösungen für medizinische Berichte können dabei auch mehrere Funktionalitäten der beschriebenen Szenarien kombinieren.
I. Transkription eines ärztlichen Diktats
In diesem Anwendungsfall diktieren die Ärztinnen und Ärzte den medizinischen Bericht nach einer vorgegebenen Struktur. Das KI-gestützte System wandelt das Diktat mithilfe von Speech-to-Text-Technologie in Text um. In manchen Fällen wird die Transkription automatisch in eine standardisierte Berichtsvorlage eingefügt, was die Konsistenz der medizinischen Dokumentation erhöht. Der fertiggestellte Bericht kann anschliessend von einer medizinischen Fachperson überprüft und freigegeben werden. Die automatische Transkription reduziert den manuellen Aufwand erheblich und spart Zeit im Arbeitsalltag. Damit eignet sich dieses Szenario besonders für niedergelassene Ärztinnen und Ärzte, und für Kliniken, die eine unkomplizierte Unterstützung bei der Dokumentation benötigen.
Eine zentrale Herausforderung liegt in der Genauigkeit der Spracherkennung, insbesondere beim Umgang mit medizinischer Fachterminologie. Obwohl eine ärztliche Nachbearbeitung vorgesehen ist, zeigen erste Praxiserfahrungen, dass dieser Schritt aus Zeitgründen teilweise ausgelassen wird. Dadurch erhöht sich das Risiko von inhaltlichen Fehlern. Eine Überprüfung durch qualifiziertes Fachpersonal – idealerweise medizinische Schreibdienste oder sogenannte Medical Scribes mit entsprechender Ausbildung – ist daher essenziell. Der Nutzen solcher Systeme liegt jedoch klar in der Effizienzsteigerung und der Verbesserung der strukturellen Qualität von Berichten.
Aus regulatorischer Sicht handelt es sich in der Regel nicht um ein Medizinprodukt, sofern die Software ausschliesslich der Transkription dient – also Sprache in Text umwandelt oder handschriftliche Notizen digitalisiert –, ohne dabei medizinische Inhalte zu analysieren und zu interpretieren oder Empfehlungen abzugeben. In diesen Fällen ist die Anwendung einem administrativen Hilfsmittel gleichzustellen. Sobald die Software jedoch medizinisch relevante Informationen verändert, etwa durch Hervorhebung bestimmter Begriffe oder automatische Zusammenfassungen, kann sie als Medizinprodukt qualifiziert werden. In solchen Fällen ist eine sorgfältige Prüfung des Verwendungszwecks und der tatsächlichen Systemfunktionen erforderlich.
II. Optimierung eines bestehenden Berichts
In diesem Anwendungsfall werden LLMs eingesetzt, um bestehende ärztliche Berichte sprachlich zu überarbeiten oder in andere Sprachen zu übersetzen. Die KI verbessert Formulierungen, optimiert den sprachlichen Stil und sorgt für eine klare, konsistente Ausdrucksweise – ohne den fachlichen Inhalt zu verändern. Besonders in internationalen oder mehrsprachigen Gesundheitseinrichtungen kann dies die Verständlichkeit und die Qualität der medizinischen Kommunikation deutlich erhöhen. Entsprechend eignet sich dieses Szenario vor allem für Spitäler, internationale Praxen und Forschungseinrichtungen, die eine effiziente sprachliche Standardisierung und Übersetzung medizinischer Dokumentationen anstreben.
Die zentrale Herausforderung besteht darin, sicherzustellen, dass durch die sprachliche Optimierung keine inhaltlichen Bedeutungsveränderungen entstehen. Auch wenn die Funktion der KI auf stilistische Anpassungen beschränkt ist, kann nicht vollständig ausgeschlossen werden, dass durch Umformulierungen auch der fachliche Gehalt beeinflusst wird. Daher ist besondere Sorgfalt bei der Überprüfung der überarbeiteten Texte geboten.
Aus regulatorischer Sicht liegt in der Regel kein Medizinprodukt vor, solange die Software ausschliesslich sprachliche oder stilistische Verbesserungen vornimmt oder Layout und Formatierung optimiert. Dieser Fall ist jedoch bereits grenzwertiger zu beurteilen als die reine Transkription, da sprachliche Anpassungen indirekt medizinische Aussagen verändern könnten. Sollte die KI hingegen eigenständig medizinische Inhalte generieren oder bestehende Diagnosen und Behandlungsvorschläge verändern, ist eine Einstufung als Medizinprodukt erforderlich. Entscheidend ist auch hier der konkrete Verwendungszweck sowie die tatsächliche Funktionsweise der Software im Anwendungskontext.
III. Erstellung eines Berichts aus der Patienteninteraktion
In diesem Anwendungsfall wird ein Gespräch zwischen einer medizinischen Fachperson und einer Patientin oder einem Patienten aufgezeichnet und mithilfe eines Speech-to-Text-Modells transkribiert und in eine strukturierte Berichtsvorlage überführt. Die sogenannte Ambient Clinical Intelligence ist insbesondere bei längeren, freien Interaktionen – etwa in der Psychiatrie oder in der hausärztlichen Versorgung – von Bedeutung. Ziel ist es, den Inhalt der Konsultation automatisch zu erfassen und für die medizinische Dokumentation nutzbar zu machen, ohne dass eine manuelle Nachbearbeitung erforderlich ist.
Der Einsatz solcher Systeme kann die Dokumentationslast erheblich reduzieren und die Arbeitsabläufe effizienter gestalten, insbesondere in Einrichtungen mit hohem Patientendurchlauf. Das KI-System muss dabei in der Lage sein, medizinisch relevante Informationen aus oft unstrukturierten, unvollständigen oder widersprüchlichen Gesprächen herauszufiltern und in eine standardisierte Form zu bringen – ohne dabei den fachlichen Inhalt zu interpretieren oder zu verändern. Genau darin liegt jedoch eine zentrale Herausforderung: In der Praxis besteht ein erhöhtes Risiko, dass LLMs Inhalte halluzinieren oder falsch gewichten. Besonders kritisch ist dies, wenn die KI nicht nur transkribiert, sondern auch Zusammenfassungen oder strukturierte Auswertungen erstellt.
Des Weiteren erfassen KI-Systeme zur Transkription primär gesprochene Inhalte, nicht jedoch klinische Beobachtungen oder vorläufige Hypothesen der medizinischen Fachperson, beispielsweise ein auffälliges Erscheinungsbild oder Verdachtsdiagnosen. Solche ergänzenden Informationen müssen weiterhin manuell dokumentiert werden, da sie wesentliche Grundlagen für die weitere medizinische Betreuung darstellen.
Aus regulatorischer Sicht gilt: Solange lediglich gesprochene Sprache in Text umgewandelt wird, handelt es sich in der Regel nicht um eine medizinische Funktion – ähnlich wie bei der reinen Transkription eines ärztlichen Diktats. Sobald das System jedoch beginnt, den Gesprächsinhalt zusammenzufassen, zu bewerten oder gar diagnostische Hinweise zu generieren, wird die Anwendung wesentlich heikler. In solchen Fällen besteht das Risiko, dass medizinisch relevante Aussagen verfälscht werden, und es ist eine klare Abgrenzung zwischen ärztlicher Entscheidung und KI-Ausgabe erforderlich. Wird die KI zur Entscheidungsunterstützung eingesetzt oder verändert sie den fachlichen Gehalt des Berichts, ist eine Einstufung als Medizinprodukt zwingend. Die Herausforderung liegt dabei nicht nur in der technischen Umsetzung, sondern auch in der rechtssicheren Kategorisierung solcher hybriden Funktionen.
IV. Bericht aus multiplen Datenquellen
In diesem Anwendungsfall kombinieren KI-gestützte Systeme unterschiedliche Datenquellen – etwa ärztliche Diktate, elektronische Patientenakten oder Gesprächsaufzeichnungen – und überführen sie in eine konsolidierte, strukturierte medizinische Dokumentation. Durch den Einsatz von LLMs können medizinische Fachpersonen auf eine integrierte Darstellung der Patientenhistorie zugreifen, was die Übersichtlichkeit verbessert und potenziell diagnostische und therapeutische Entscheidungen unterstützt.
Die Chancen solcher Systeme liegen in der Effizienzsteigerung und im besseren Zugang zu relevanten Informationen über verschiedene Dokumentationsformen hinweg. Gleichzeitig bestehen erhebliche Herausforderungen in der zuverlässigen Zusammenführung und Interpretation der Inhalte. Insbesondere widersprüchliche Diagnosen, mögliche unvollständige Angaben oder mehrdeutige Formulierungen aus unterschiedlichen Quellen können zu fehlerhaften oder irreführenden Ausgaben führen. Es ist daher entscheidend, dass KI-Systeme Herkunft, Kontext und Bedeutung der Informationen korrekt zuordnen und transparent darstellen, damit medizinisches Fachpersonal die Inhalte überprüfen kann. Um die medizinische Relevanz und die Richtigkeit der generierten Informationen fundiert beurteilen zu können, braucht es ausreichend klinische Erfahrung.
Aus regulatorischer Sicht ist zu unterscheiden: Solange die Software ausschliesslich vorhandene Informationen aggregiert, in strukturierter Form darstellt und ihre Herkunft nachvollziehbar macht – ohne sie medizinisch zu bewerten oder zu verändern –, handelt es sich vermutlich nicht um ein Medizinprodukt. Wenn das System jedoch Daten analysiert, etwa durch Trendanalysen, oder gar eigenständig medizinische Schlussfolgerungen zieht, Diagnosen ableitet oder Behandlungsempfehlungen formuliert, greift der medizinische Verwendungszweck. In solchen Fällen ist eine Einstufung als Medizinprodukt erforderlich, verbunden mit den entsprechenden Anforderungen an Sicherheit, Nachvollziehbarkeit und klinische Validierung.
V. Chatbot für die Patientenakte
In diesem Anwendungsfall ermöglicht ein KI-gestützter Chatbot medizinischem Fachpersonal, gezielt Informationen aus der Patientenakte abzurufen – etwa um relevante Befunde zu identifizieren, Zusammenhänge zu erkennen oder Differenzialdiagnosen in Betracht zu ziehen. Die Abfrage kann zudem um medizinische Leitlinien und Standards ergänzt werden, was eine evidenzbasierte Entscheidungsfindung unterstützt und zur Standardisierung der Behandlung beitragen kann. Durch die zeitnahe Bereitstellung relevanter Informationen lassen sich klinische Entscheidungen effizienter treffen, was insbesondere in zeitkritischen Situationen die Versorgungsqualität verbessern kann.
Die Herausforderung liegt in der sinnvollen Verknüpfung und Interpretation verschiedenartiger Datenquellen. Wie bei anderen Szenarien mit multiplen Datenquellen können auch hier widersprüchliche, unvollständige oder mehrdeutige Informationen auftreten, die die Aussagekraft des Chatbots beeinträchtigen. Je besser das System in der Lage ist, komplexe Informationen zusammenzuführen und klinisch relevante Antworten zu formulieren, desto höher ist sein Nutzen – aber auch sein Risiko, insbesondere wenn es in sicherheitskritischen Situationen zum Einsatz kommt, etwa zur Identifikation von Kontraindikationen vor einer Medikamentengabe.

Aus regulatorischer Sicht ist entscheidend, wie das System eingesetzt wird und welchen Zweck es erfüllt. Handelt es sich lediglich um eine kontextsensitive Suche, die bestehende Inhalte wie Dokumentenauszüge oder Leitlinientexte auffindbar macht und wiedergibt, ohne sie zu verändern oder neu zu bewerten, liegt in der Regel kein Medizinprodukt vor. Wird das System jedoch so eingesetzt, dass es Informationen verarbeitet und in aufbereiteter Form bereitstellt, um ärztliche Entscheidungen aktiv zu unterstützen – etwa durch die Formulierung möglicher Diagnosen oder Therapieempfehlungen –, handelt es sich um ein Medizinprodukt. In solchen Fällen sind je nach Einsatzbereich auch höhere Anforderungen an Sicherheit, Nachvollziehbarkeit und klinische Validierung zu erfüllen.
VI. KI-gestützte Vorschläge für Differenzialdiagnosen
In diesem Anwendungsfall wird ein ärztliches Diktat zunächst mittels eines Speech-to-Text-Modells transkribiert und in eine strukturierte Berichtsvorlage überführt. Anschliessend analysiert ein LLM die erfassten Informationen – etwa Symptome, anamnestische Angaben, Laborbefunde oder andere relevante Daten – und generiert daraufhin eine Liste möglicher Differenzialdiagnosen. Diese werden dem medizinischen Fachpersonal zur weiteren Prüfung, Ergänzung oder Verwerfung angezeigt. Die finale Diagnoseentscheidung bleibt ausdrücklich bei den behandelnden Ärztinnen und Ärzten.
Solche Systeme können die diagnostische Entscheidungsfindung sinnvoll unterstützen, indem sie relevante, aber nicht sofort offensichtliche Alternativen aufzeigen und so die diagnostische Sicherheit erhöhen. Sie können zudem helfen, seltene oder schwer erkennbare Erkrankungen frühzeitig zu berücksichtigen, und auf Basis strukturierter Informationen klinisch fundierte Vorschläge generieren. Besonders in diagnostisch komplexen Fachbereichen – etwa in der inneren Medizin oder in der Notfallaufnahme – bietet diese Technologie das Potenzial, fundierte Einschätzungen schneller zu ermöglichen. Gleichzeitig besteht jedoch ein erhöhtes Risiko von Fehlinformationen oder sogenannten Halluzinationen durch das Sprachmodell. Daher ist es essenziell, dass die KI-Ausgaben klar als Vorschläge gekennzeichnet sind und die ärztliche Kontrolle jederzeit gewährleistet bleibt.
Aus regulatorischer Sicht liegt bei diesem Szenario eindeutig eine medizinische Entscheidungsunterstützung vor. Die KI greift direkt in den diagnostischen Entscheidungsprozess ein, indem sie Diagnosen generiert und teilweise auch priorisiert. Damit ist der medizinische Verwendungszweck klar gegeben, was gemäss Definition zu einer Einstufung als Medizinprodukt führt – voraussichtlich in der Risikoklasse IIa oder höher, abhängig vom klinischen Anwendungsfeld und von den potenziellen Risiken im Fall einer Fehlfunktion. Für solche Systeme gelten erhöhte Anforderungen an Sicherheit, Transparenz, Nachvollziehbarkeit und klinische Bewertung. Die Funktionsweise des Modells muss technisch dokumentiert und transparent nachvollziehbar sein – einschliesslich der verwendeten Datenquellen, Validierungsmethoden und Metriken für Genauigkeit, Robustheit und Zuverlässigkeit.
Obwohl bestehende regulatorische Rahmenwerke wie die MDR oder ISO 82304-1 technologieoffen gestaltet sind und grundsätzlich Produkte und nicht Technologien regeln, stellt die Anwendung auf LLM-basierte Systeme besondere Herausforderungen dar. Aufgrund ihrer Dynamik, ihrer geringen Nachvollziehbarkeit und ihrer potenziellen Anpassungsfähigkeit ist von einem erhöhten Dokumentationsaufwand sowie von einer komplexeren Zulassungsprüfung auszugehen. Der Einsatz solcher Systeme ist vielversprechend, muss jedoch mit besonderer Vorsicht erfolgen. Eine klinische Validierung, eine klare Abgrenzung zwischen ärztlicher Entscheidung und KI-Unterstützung sowie eine transparente Kommunikation gegenüber den Nutzenden zum Einsatz von KI-Systemen sind entscheidend, um sowohl den rechtlichen Anforderungen zu genügen als auch die Patientensicherheit zu gewährleisten. Erste wissenschaftliche Studien unterstreichen das Potenzial dieser Technologien bei der Unterstützung von Differenzialdiagnosen.
VII. Automatisierte Abrechnung
In diesem Anwendungsfall erstellt eine KI-gestützte Software auf Basis eines medizinischen Berichts – und gegebenenfalls weiterer Datenquellen wie etwa Sprechstundenaufzeichnungen – automatisch eine Leistungsabrechnung. Besonders grössere Gemeinschaftspraxen und Spitäler können durch den Einsatz von LLMs die Abrechnungsprozesse vereinheitlichen und so Diskrepanzen zwischen den einzelnen Leistungserbringenden reduzieren. Ein typisches Beispiel ist die automatisierte Zuordnung eines passenden ICD-Codes zu einer gestellten Diagnose, um eine konsistente Dokumentation und Abrechnung zu unterstützen.
Die Vorteile liegen vor allem in der Effizienzsteigerung und der Standardisierung administrativer Abläufe. Gleichzeitig besteht die Herausforderung darin, dass auch rein organisatorische Prozesse in einen medizinisch relevanten Bereich übergehen können – insbesondere wenn die KI nicht nur Informationen verarbeitet, sondern diese auch auswertet und auf ihre medizinische Relevanz hin beurteilt.
Aus regulatorischer Sicht gilt eine Software zur automatisierten Abrechnung grundsätzlich als nicht medizinisch, da sie primär organisatorischen oder finanziellen Zwecken dient. Wird die Software jedoch eingesetzt, um etwa auf Basis einer Diagnose die medizinische Rechtfertigung einer Behandlung zu prüfen oder Behandlungsentscheide zu beeinflussen, kann dies eine medizinische Zweckbestimmung darstellen. In solchen Fällen muss sorgfältig geprüft werden, ob die Anwendung unter die Medizinprodukteverordnung fällt. Besonders relevant ist die sogenannte Sekundärnutzung: Auch wenn eine Diagnose durch KI ursprünglich nur für Abrechnungszwecke erstellt wird, kann eine medizinische Zweckbestimmung entstehen – etwa wenn die Information ohne ärztliche Validierung in medizinische Berichte einfliesst und dadurch Behandlungsentscheidungen beeinflusst werden.
VIII. Automatisches Aufgebot von Patientinnen und Patienten
Beim automatischen Aufgebot handelt es sich um eine KI-basierte Anwendung, bei der Patientinnen und Patienten basierend auf vordefinierten Kriterien, medizinischen Berichten, Patientenakten und Terminplänen automatisch zu einem Untersuchungstermin eingeladen werden. Die KI unterstützt dabei die Terminverwaltung und trägt zur effizienten Auslastung vorhandener Ressourcen bei, insbesondere in stark frequentierten Praxen oder Spitälern. Ziel ist es, administrative Prozesse zu entlasten und die Versorgungsplanung zu optimieren. Die Herausforderung bei diesem Einsatz besteht in der Unterscheidung zwischen rein administrativen Abläufen und medizinisch motivierten Entscheidungen. Wird das Aufgebot beispielsweise aufgrund fixer Kontrollintervalle oder bekannter Therapieschemata generiert, bleibt die Funktion im administrativen Bereich. Kritischer wird es, wenn die KI aufgrund der Auswertung medizinischer Inhalte – etwa durch Erkennen einer Auffälligkeit in einem Bericht – eigenständig entscheidet, ob und wann eine Patientin oder ein Patient aufgeboten werden soll. In diesem Fall liegt eine medizinische Entscheidungsunterstützung vor.
Aus regulatorischer Sicht ist daher klar zu differenzieren: Erfolgt die Terminvergabe automatisiert, aber ausschliesslich auf Basis administrativer Parameter, handelt es sich nicht um ein Medizinprodukt. Sobald jedoch die Entscheidung über das Aufgebot direkt auf einer medizinischen Analyse basiert – etwa weil die KI aus einem Bericht Handlungsbedarf ableitet –, ist von einer medizinischen Zweckbestimmung auszugehen. In solchen Fällen kann eine Einstufung als Medizinprodukt erforderlich werden, verbunden mit entsprechenden Anforderungen an Sicherheit, Nachvollziehbarkeit und klinische Bewertung.
Regulatorische Einordnung medizinischer Software
Die aufgezeigten Anwendungsfälle verdeutlichen, wie eng die Einstufung einer Software als Medizinprodukt mit ihrer konkreten Zweckbestimmung und Funktionalität verknüpft ist. Sobald eine Software medizinische Inhalte analysiert, interpretiert oder potenziell Entscheidungsprozesse beeinflusst, unterliegt sie den Anforderungen der Medizinprodukteverordnung. Umso wichtiger sind daher eine technisch saubere Umsetzung, transparente Funktionsbeschreibungen sowie eine klare Trennung zwischen administrativen Funktionen und medizinischer Entscheidungsunterstützung – als Grundlage für regulatorische Konformität und Patientensicherheit. Gerade bei Anwendungen, deren Qualifikation nicht ganz klar ist, sollte man sich von regulatorischen Beratungsunternehmen Unterstützung holen und z.B. eine Regulatory Opinion erstellen lassen.
Verwenden Sie die Akkordeon-Bedienelemente, um die Sichtbarkeit der jeweiligen Panels (unterhalb der Bedienelemente) umzuschalten.
Medizinprodukte sind in einem ersten Schritt zu klassifizieren. Die Einteilung erfolgt gemäss Anhang VIII MDR in vier verschiedene Klassen, anhand des vom Produkt ausgehenden Risikos, das stark vom Zustand der Patientinnen und Patienten abhängt (I, IIa, IIb, III). Relevant ist ferner, wie signifikant die Informationen sind, die die Software liefert. Hier gibt es drei Möglichkeiten: hoch (entscheidet direkt über Behandlung und Diagnose), mittel (beeinflusst das klinische Management massgeblich) oder gering (informiert lediglich das klinische Management).
Eine genauere Auflistung der Klassifizierungsregeln mit Beispielen findet sich im Leitfaden MDCG 2019-11 oder im Borderline-Manual der EU. Gemäss Klassifizierungsregel 11 in Anhang VIII MDR fallen nur Produkte in die Klasse I, die keine Informationen liefern, die für diagnostische oder therapeutische Zwecke genutzt werden können. Die folgende Tabelle aus Anhang III des MDCG 2019-11 zeigt, wie sich der Gesetzgeber die Klassifizierung einer diagnostischen oder therapeutischen Software vorstellt.
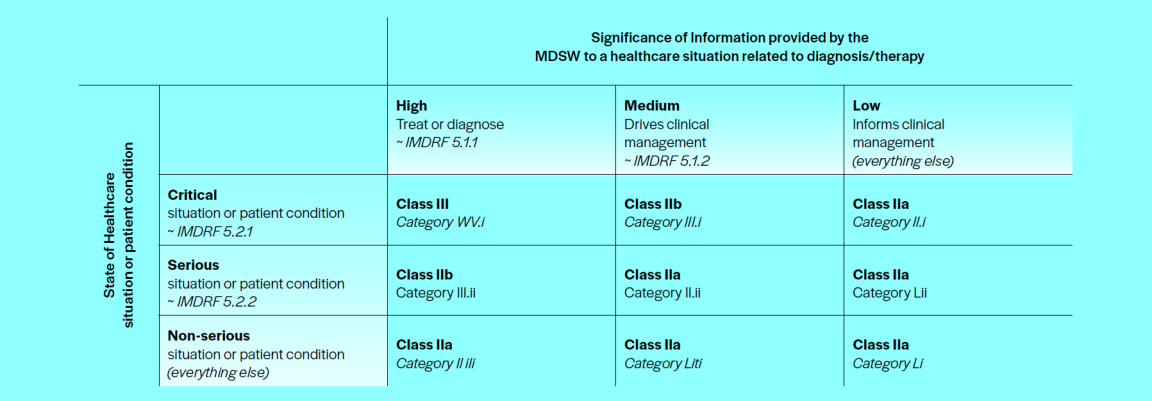
Wird eine Software nach den geltenden Bestimmungen als Produkt nach MepV oder IvDV qualifiziert, so müssen diverse Regularien und Normen berücksichtigt werden. Dabei ist es wichtig, zwischen Systemnormen (Anforderungen an das Unternehmen oder die Organisation) und Produktnormen (Anforderungen an das Medizinprodukt selbst) zu unterscheiden.
|
Wesentliche Systemnorm
|
|
|---|---|
|
SN EN ISO 13485
|
Medizinprodukte – Qualitätsmanagementsysteme – Anforderungen für regulatorische Zwecke |
|
Wesentliche Produktnormen
|
|
|---|---|
|
SN EN 62304
|
Medizingeräte-Software – Software-Lebenszyklus-Prozesse |
|
SN EN 82304-1
|
Gesundheitssoftware – Teil 1: Allgemeine Anforderungen für die Produktsicherheit |
|
SN EN 62366 – 1
|
Medizinprodukte – Teil 1: Anwendung der Gebrauchstauglichkeit auf Medizinprodukte |
Die Zertifizierung des Herstellers nach ISO 13485 ist zwingend erforderlich, falls das Produkt der Klasse IIa oder höher (bzw. der IvDV-Klasse B oder höher) zugeteilt wird. Es existiert jedoch eine Vielzahl weiterer Normen, deren Prüfung vom Kontext der jeweiligen Software abhängt. Insbesondere im Bereich des maschinellen Lernens sind momentan einige neue Normen von IEC und ISO geplant, die auch in der Schweiz Anwendung finden werden.
Angesichts der zunehmenden Cyberangriffe und der wachsenden Bedeutung von Cybersicherheit ist eine Zertifizierung des Produktherstellers nach der ISO/IEC-27000-Normenreihe (z.B. ISO/IEC 27001 für das Informationssicherheitsmanagement) empfehlenswert. Diese Zertifizierung betrifft nicht das Medizinprodukt selbst, sondern das Sicherheitsmanagement des Herstellers – insbesondere bei cloudbasierten oder vernetzten Anwendungen. Alternativ kann man sich als produktbezogene Ergänzung an der Norm IEC 81001-5-1 orientieren, die auch von europäischen Zulassungsstellen anerkannt wird.
Wer in der Schweiz ein Medizinprodukt in Verkehr bringt, muss in erster Linie die allgemeinen Sicherheits- und Leistungsanforderungen (GSPR) gewährleisten, die sich in Anhang I der MDR finden. Der Nachweis, dass die GSPR eingehalten werden, umfasst eine klinische Bewertung und eine Risikoanalyse, welche die Sicherheit und die Leistungsfähigkeit des Produkts belegen. Zudem ist eine technische Dokumentation zu erstellen, in der die Angaben nach den Anhängen II und III der EU-MDR bzw. EUIVDR aufzuführen sind.
KI-Anbieter müssen auch eine Konformitätsbewertung vornehmen. In wenigen Fällen genügt eine Konformitätsbewertung des Herstellers (Software der Klasse I ohne Messfunktion), in allen übrigen Fällen muss eine Konformitätsbewertung durch eine bezeichnete Stelle erfolgen. Die Produkte müssen sodann das Konformitätskennzeichen tragen.
Seit der Aufkündigung der gegenseitigen Anerkennung von Medizinprodukten der EU im Jahr 2021 kann man Produkte der Klasse IIa und höher (bzw. der IvDV-Klasse B und höher) nicht mehr in der Schweiz zertifizieren lassen. Man benötigt dafür eine Zulassungsstelle in der EU, eine sogenannte benannte Stelle. Die Schweiz anerkennt unilateral die EU-Zulassung als Medizinprodukt.
Besondere und erleichterte Anforderungen gelten für Medizinprodukte, die in Gesundheitseinrichtungen hergestellt und verwendet werden (Art. 9 MepV und 9 IvDV). Grundsätzlich gilt eine solche Software als in Betrieb genommen, und die einschlägigen grundlegenden Sicherheits- und Leistungsanforderungen sind ohne Einschränkung zu erfüllen. Jedoch gelten die übrigen Anforderungen nach MepV bzw. IvDV nicht. Nähere Informationen dazu findet man im Leitfaden MDCG 2023-1. Gesundheitseinrichtungen müssen die hergestellten und verwendeten Medizinprodukte vor der Inbetriebnahme melden (Art. 18 MepV und Art. 10 IvDV).
Aus Haftungssicht ist es sehr zu empfehlen, keine generischen LLMs in einem Medizinprodukt zu verwenden, da die Hersteller dieser LLMs in ihren AGB professionelle Gesundheitsanwendungen meist ausschliessen (z.B. OpenAI Usage Policy). Als Anbieter einer LLM-gestützten Gesundheitslösung riskiert man so, für Falschaussagen des LLM haftbar zu werden. Es existieren aber inzwischen eine ganze Reihe von LLMs, die spezifisch mit Medizindaten trainiert wurden und die auch für medizinische Anwendungen vorgesehen sind (z.B. Med-PaLM 2 oder BioGPT). Eine Schweizer Alternative, die sogar open source verfügbar ist, ist Meditron von der EPFL. Anbieter können auf diesem Grundlagensystem aufbauen und durch zusätzliche technische sowie organisatorische Massnahmen die Genauigkeit und Zuverlässigkeit des LLM im jeweiligen Anwendungskontext gezielt verbessern. Ergänzend kann der Einsatz von Retrieval-Augmented Generation dazu beitragen, medizinisches Fachwissen kontrolliert einzubinden und Halluzinationen sowie kontextbezogene Antworten zu reduzieren.
Primär ist der Hersteller verantwortlich für die Qualifizierung und Klassifizierung seiner Software als Medizinprodukt sowie für ihre Konformität, ihre Sicherheit und ihre Leistungsfähigkeit. Darüber hinaus tragen auch andere Akteure entlang des Lebenszyklus Verantwortung: Importeure und Händler müssen sicherstellen, dass nur konforme Produkte in Verkehr gebracht werden und entsprechende Kontrollen und Dokumentationspflichten erfüllen. Betreiber wie Krankenhäuser oder Arztpraxen sind verpflichtet, eine ordnungsgemässe Installation, Nutzung und Wartung sicherzustellen sowie Vorkommnisse zu melden. Patientinnen und Patienten haben keine direkten Pflichten, jedoch ein Recht auf sichere und wirksame Produkte. Besondere Herausforderungen ergeben sich bei KI-Systemen mit kontinuierlichem Lernverhalten (Continuous Learning), da sich deren Verhalten nach dem Inverkehrbringen verändert. Eine vollständige Ex-ante-Bewertung von Sicherheit und Leistungsfähigkeit ist daher nur begrenzt möglich und erfordert ergänzende Kontrollmechanismen im laufenden Betrieb (Predetermined Change Control Plans, siehe Kapitel 5.1., Punkt 9).
«LLMs passen in bestehende regulatorische Konzepte – vorausgesetzt die Funktionsabgrenzung ist klar und die Dokumentation sorgfältig.»
Dr. med. Atanas Todarov, Chief Medical Officer, Arcondis
Bitte geben Sie uns Feedback
Ist diese Seite verständlich?
Vielen Dank für Ihr Feedback!
Kontakt
Amt für Wirtschaft – Standortförderung
Montag bis Freitag
8.00 bis 12.00 Uhr und
13.30 bis 17.00 Uhr
